![]()
Use mruby to evaluate expressions in samtools!
git clone https://github.com/kojix2/samtools-mruby
make
samtools/samtools tanuki # check if it works __,-─-、__
(〆-─-ヽ)
( ´・ω・` )
/ ,r‐‐‐、ヽ
し l x )J
_.'、 ヽ ノ.人
(_((__,ノU´U. (酒)
Tanuki in mruby (3.3.0)
Rake is required to build mruby.
If you are using conda to install Ruby, set the LD environment variable:
rake LD=/usr/bin/gcc MRUBY_CONFIG=$(pwd)/mruby_build_config.rb -f $(pwd)/mruby/RakefileThe samtools-mruby project allows you to use mruby expressions to manipulate and analyze BAM files. The available variables include:
endpos: Alignment end position (1-based)flags: Combined FLAG fieldpaired,proper_pair,unmap,munmap,reverse,mreverse,read1,read2,secondary,qcfail,dup,supplementary- These can be used with or without the
?suffix, e.g.,pairedandpaired?are equivalent.
hclen: Number of hard clipped baseslibrary: Library (LB header via RG)mapq: Mapping qualitympos: Synonym for pnextmrefid: Mate reference number (0 based)mrname: Synonym for rnextncigar: Number of CIGAR operationspnext: Mate's alignment position (1-based)pos: Alignment position (1-based)qlen: Alignment length: no. query basesqname: Query namequal: Quality values (raw, 0-based)refid: Integer reference number (0 based)rlen: Alignment length: no. reference basesrname: Reference namernext: Mate's reference namesclen: Number of soft clipped basesseq: Sequencetlen: Template length (insert size)tag: XX tag value
These variables enable detailed data manipulation and analysis.
-
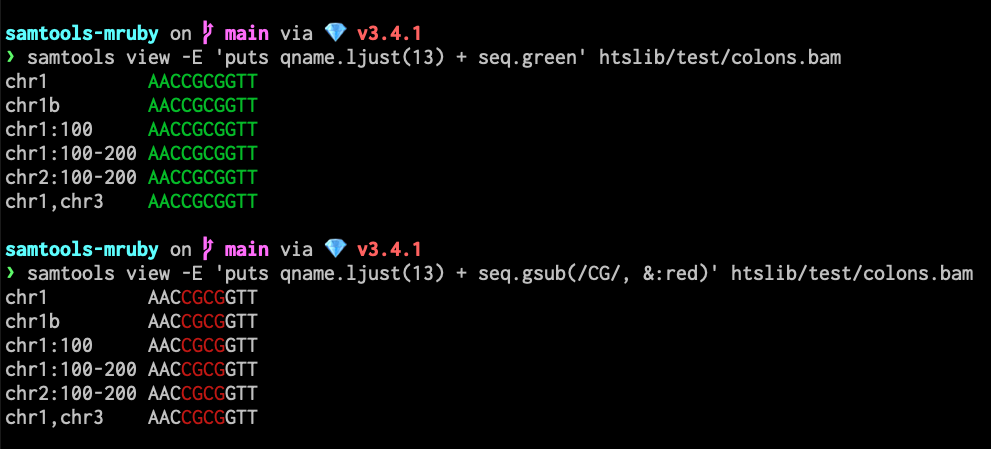
Basic Usage: Output read name and sequence in green.
samtools view -E 'puts qname.ljust(13) + seq.green' htslib/test/colons.bam -
Pattern Highlighting: Use regular expressions.
samtools view -E 'puts qname.ljust(13) + seq.gsub(/CG/, &:red)' htslib/test/colons.bam -
Flag Methods: Access SAM flag information. Flag methods can be used with or without the
?suffix. For example,pairedandpaired?are equivalent.samtools view -E 'puts "#{qname} is paired" if paired?' example.bam -
Tag Access: Retrieve BAM tags.
samtools view -E 'puts "NM:#{tag("NM")}" if tag("NM")' example.bam -
Custom Filtering: Use expressions for filtering.
samtools view -E 'puts qname if prpper_pair?' example.bam # samtools view -E 'puts qname if flags & 0x2 != 0' example.bam
- Local: Defined inside expressions, do not persist.
- Global: Defined with
$, persist across records.
Example to count mapped reads:
samtools view -E '$count ||= 0; $count += 1 unless unmap?; END { puts $count }' example.bamThe samtools-mruby project integrates mruby into samtools, allowing for enhanced functionality through mruby expressions. This integration includes:
- Makefile Modifications: Added support for mruby by including
MRBDIR,MRB_CPPFLAGS, andMRB_LDFLAGS. Updated object files list to includetanuki.oandsam_view_mruby.o. - bamtk.c: Introduced a new
tanukicommand, which displays a tanuki using mruby. - sam_view.c: Added support for evaluating mruby expressions with a new
mruby_exprfield in settings. Integrated mruby initialization and finalization. - New Files:
sam_view_mruby.c: Implements methods for interacting with BAM records using mruby.sam_view_mruby.h: Header file forsam_view_mruby.c.tanuki.c: Contains the implementation for thetanukicommand.
To see changes made to the original samtools repository:
git -C samtools diff origin/develop...origin/mruby- The
mrubyandhtslibdirectories are submodules. samtoolsis based on themrubybranch of the kojix2 repository.- The
tanukisubcommand distinguishes between standard and mruby-enhanced samtools.
Send pull requests to the mruby branch of my samtools repository.
- MIT License
- This tool was created actively using code generators such as ChatGPT and Copilot.