
Paper | Tutorials | Examples | Demo
{kind=link}
MolScore contains code to score de novo compounds in the context of generative de novo design by generative models via the subpackage molscore, as well as, facilitate downstream evaluation via the subpackage moleval. An objective is defined via a JSON file which can be shared to propose new multi-parameter objectives for drug design. MolScore can be used in several ways:
- To implement a multi-parameter objective to for prospective drug design.
- To benchmark objectives/generative models/optimization using benchmark mode (MolScoreBenchmark).
- To implement a sequence of objectives using curriculum mode (MolScoreCurriculum).
Contributions and/or ideas for added functionality are welcomed!
Install MolScore with PyPI (recommended):
pip install molscore --upgrade
or directly from GitHub:
git clone https://github.com/MorganCThomas/MolScore.git
cd MolScore ; pip install -e .
Note: I recommend mamba for environment handling
Simplest integration of MolScore requires a config file, for example:
from molscore import MolScore
ms = MolScore(
model_name='test',
task_config='molscore/configs/QED.json',
budget=10000
)
while not ms.finished:
scores = ms.score(SMILES)Note: see tutorial for more detail
A GUI exists to help write the config file, which can be run with the following command.
molscore_config
Note: see tutorial for more detail
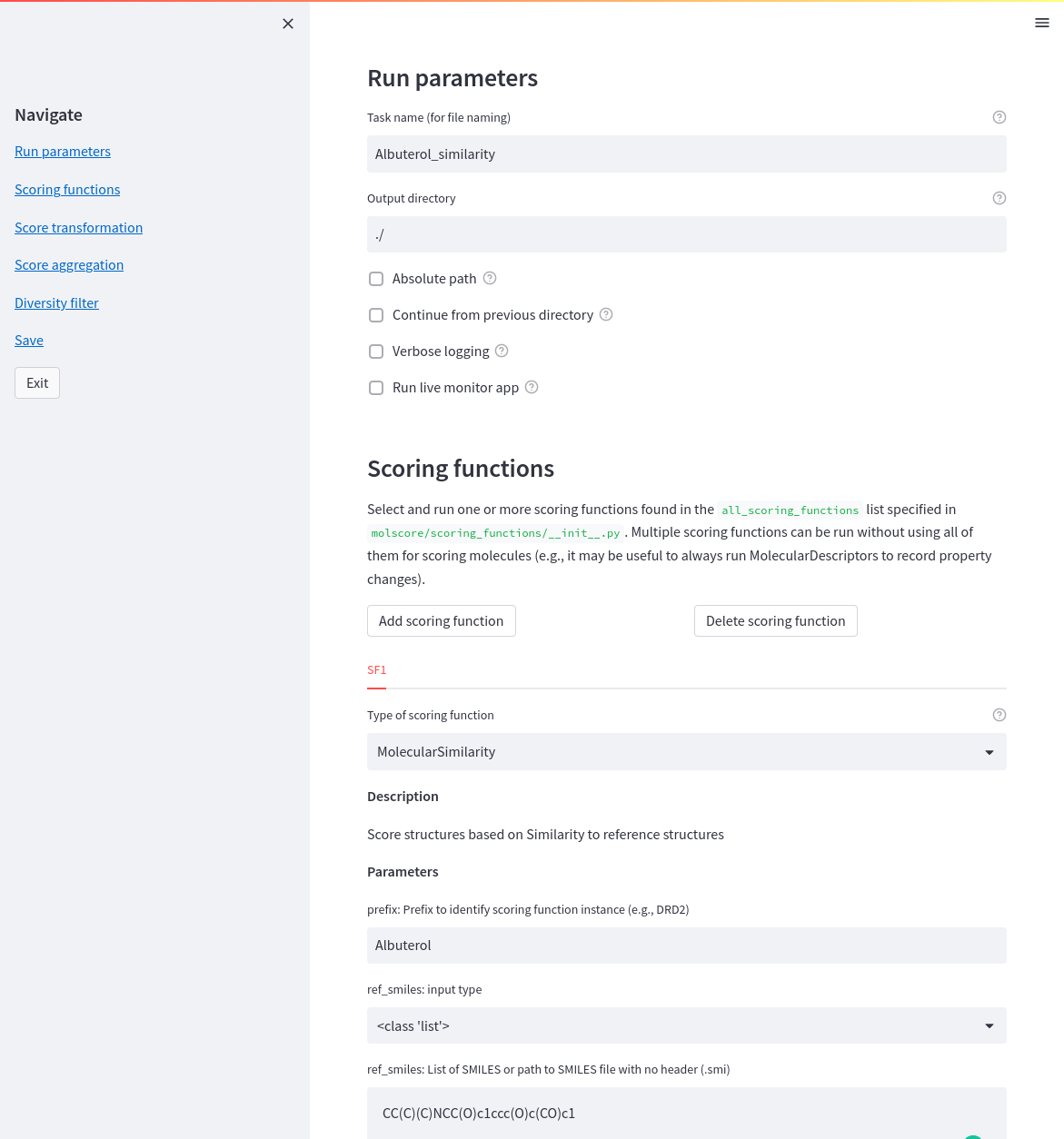
Scoring functionality
Scoring functions
- Descriptors: RDKit, Maximum consecutive rotatable bonds, Penalized LogP, LinkerDescriptors (Fragment linking),
- MolSkill: Extracting medicinal chemistry intuition via preference machine learning as available on Nature Communications.
- Synthesizability: RAscore, AiZynthFinder, SAscore, ReactionFilters (Scaffold decoration)
- 2D Similarity: Fingerprint similarity (any RDKit fingerprint and similarity measure), substructure match/filter, Applicability domain
- 3D Similarity: ROCS, Open3DAlign
- QSAR: Scikit-learn (classification/regression), ChemProp
- Docking: Glidea, Smina, OpenEyea, GOLDa, PLANTS, rDock, Vina, Gnina
- Ligand preparation: RDKit->Epik, Moka->Corina, Ligprep, Gypsum-DL
a Requires a license
Transformation functions (transform values to [0-1])
- Linear
- Linear threshold
- Step
- Step threshold
- Gaussian
Aggregation functions (combine multiple scores into 1)
- Arithmetic mean
- Geometric mean
- Weighted sum
- Weighted product
- Auto-weighted sum/product
- Pareto front
Filters (applied to final aggregated score)
- Any scoring function as a filter
- Diversity filters
Benchmarks are lists of objectives (configuration files) with metrics calculated upon exit. Re-implementations of existing benchmarks are available as presets.
from molscore import MolScoreBenchmark
msb = MolScoreBenchmark(
model_name='test',
output_dir='./',
benchmark='GuacaMol',
budget=10000
)
for task in msb:
while not task.finished:
scores = task.score(SMILES)Current benchmarks available include: GuacaMol, GuacaMol_Scaffold, MolOpt, 5HT2A_PhysChem, 5HT2A_Selectivity, '5HT2A_Docking', LibINVENT Exp1&3, MolExp(L)
Note: inspect preset benchmarks with
MolScoreBenchmark.presets.keys()
Note: see tutorial for more detail
The moleval subpackage can be used to calculate metrics for an arbitrary set of molecules.
from moleval.metrics.metrics import GetMetrics
MetricEngine = GetMetrics(
test=TEST_SMILES, # Model training data subset
train=TRAIN_SMILES, # Model training data
target=TARGET_SMILES, # Exemplary target data
)
metrics = MetricEngine.calculate(
GEN_SMILES, # Generated data
)Note: see tutorial for more detail
Metrics available
Intrinsice metrics (generated molecules only)
- Validity, Uniqueness, Scaffold uniqueness, Internal diversity (1 & 2), Scaffold diversity
- Sphere exclusion diversity: Measure of chemical space coverage at a specific Tanimoto similarity threshold. I.e., A score 0.5 indicates 50% of the sample size sufficiently describes the chemical space, therefore the higher the metric the more diverse the sample. Also see here
- Solow Polasky diversity
- Functional group diversity
- Ring system diversity
- Filters: Passing of a set of drug-like filters (MolWt, Rotatable bonds, LogP etc.), Medicinal Chemistry substructures and PAINS substructures.
- Purchasability: Molbloom prediction of presence in ZINC20
Extrinsic metrics (comparison to reference molecules)
- Novelty
- FCD
- Analogue similarity: Proportion of generated molecules that are analogues to molecules in reference data.
- Analogue coverage: Proportion of reference data that are analogues to generated data.
- Functional group similarity
- Ring system similarity
- Single nearest neighbour similarity
- Fragment similarity
- Scaffold similarity
- Outlier bits (Silliness): Average proportion of fingerprint bits (atomic environments) present in a generated molecule, not present anywhere in the reference data. The lower the silliness the better.
- Wasserstein distance (LogP, SA Score, NP score, QED, Weight)
-
Curriculum learning (see tutorial)
-
Experience replay buffers (see tutorial)
-
Parallelisation (see tutorial)
-
A GUI for monitoring generated molecules (see below)
molscore_monitor

If you use this software, please cite it as below.
@article{thomas2024molscore,
title={MolScore: a scoring, evaluation and benchmarking framework for generative models in de novo drug design},
author={Thomas, Morgan and O’Boyle, Noel M and Bender, Andreas and De Graaf, Chris},
journal={Journal of Cheminformatics},
volume={16},
year={2024},
publisher={BMC}
}
This software was also utilised in the following publications:
- Thomas, M., Smith, R.T., O’Boyle, N.M. et al. Comparison of structure- and ligand-based scoring functions for deep generative models: a GPCR case study. J Cheminform 13, 39 (2021). https://doi.org/10.1186/s13321-021-00516-0
- Thomas M, O'Boyle NM, Bender A, de Graaf C. Augmented Hill-Climb increases reinforcement learning efficiency for language-based de novo molecule generation. J Cheminform 14, 68 (2022). https://doi.org/10.1186/s13321-022-00646-z
- Handa K, Thomas M, Kageyama M, Iijima T, Bender A. On the difficulty of validating molecular generative models realistically: a case study on public and proprietary data. J Cheminform 15, 112 (2023). https://doi.org/10.1186/s13321-023-00781-1
- Thomas M, Ahmad M, Tresadern G, de Fabritiis G. PromptSMILES: Prompting for scaffold decoration and fragment linking in chemical language models. J Cheminform 16, 77 (2024). https://doi.org/10.1186/s13321-024-00866-5
- Bou A, Thomas M, Dittert S, Ramírez CN, Majewski M, Wang Y, Patel S, Tresadern G, Ahmad M, Moens V, Sherman W. ACEGEN: Reinforcement learning of generative chemical agents for drug discovery. J Chem Inf Model 64, 15 (2024). https://doi.org/10.1021/acs.jcim.4c00895
- Thomas M, Matricon PG, Gillespie RJ, Napiórkowska M, Neale H, Mason JS, Brown J, Fieldhouse C, Swain NA, Geng T, O'Boyle NM. Identification of nanomolar adenosine A2A receptor ligands using reinforcement learning and structure-based drug design. Nat Commun 16, 5485 (2025). https://doi.org/10.1038/s41467-025-60629-0